
Type I (Non-neuronopathic) Gaucher’s disease with bone marrow involvement: a rare case report.
Keywords:
Gaucher’s disease, Glucocerebrosidase, Non-neuronopathic
Abstract
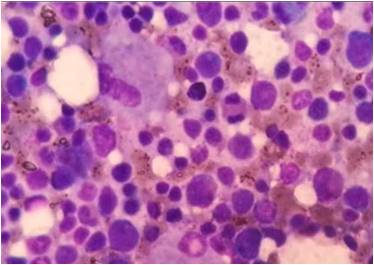
Gaucher’s disease an uncommon autosomal recessive sphingolipid lysosomal storage disease characterized by the deposition of glucocerebroside in cells of the macrophage-monocyte system from the deficient activity of the lysosomal hydrolase β-glucosidase( glucocerebrosidase). In the absence of enzyme, glucosylceramide accumulates in lysosomes of macrophages of tissues with multisystem organ involvement viz. liver, spleen, bone marrow, lungs and central nervous system.Serum β glucosidase levels <15% of mean normal activity confirms the diagnosis, enzyme replacement being the only definitive treatment. We report this case of 1 year 6 month male child who presented with delayed milestones, purpuric rashes and massive hepatosplenomegaly. Hematological workup revealed pancytopenia. Bone marrow study showed the characteristic gaucher cells and liver biopsy also showed evidence of Gauchers disease. Hence diagnosis of Type I (non-neuronopathic) Gaucher disease was given. With the advent of enzyme replacement therapy and substrate reduction therapy the natural history of the disease has been changed with a marked decrease in morbidity. The incidence of Type I Gaucher’s disease is 1 in 60,000 births but this non-neuronopathic type presenting with pancytopenia due to bone marrow involvement is rare entity with very few cases in India.References
1. Shehi B, Boçari G, Vyshka G, Xhepa R , Alushani D. Gaucher’s Disease in Albanian Children: Casuistics and Treatment. Iranian Journal of Pediatrics 2011; 21( 1): 1-7.
2. Epstein E. Beitrag zurchemie der Gaucherschen krankeit. Biochem Z. 1924;145:398–402.
3. Jain VV, Yelwatkar S. Unusual presentation of adult Gaucher’s disease: A long and difficult road to diagnosis.Indian J Endocr Metab 2011;15:224-6.
4.Nagral A. Gaucher Disease. J Clin Exp Hepatol. 2014 Mar; 4(1): 37–50.
5.Schnabel D, Schroder M, Sandhoff K. Mutation in the sphingolipid activator protein 2 in a patient with a variant of Gaucher disease. FEBS Lett. 1991;284:57–9.
6.Ho M.W., Seck J., Schmidt D. Adult Gaucher's disease: kindred studies and demonstration of a deficiency of acid beta-glucosidase in cultured fibroblasts. Am J Hum Genet. 1972;24:37–45.
7.Giraldo P., Cenarro A., Alfonso P. Chitotriosidase genotype and plasma activity in patients type 1 Gaucher's disease and their relatives (carriers and non carriers) Hematologica. 2001;86:977–984.
8. Svennerholm L., Håkansson G., Lindsten J., Wahlström J., Dreborg S. Prenatal diagnosis of Gaucher disease. Assay of the beta-glucosidase activity in amniotic fluid cells cultivated in two laboratories with different cultivation conditions. Clin Genet. 1981;19:16–22.
9.Di Rocco M, Giona F, Carubbi F, Linari S, Minichilli F, Brady RO, et al. A new severity score index for phenotypic classification and evaluation of responses to treatment in type I Gaucher disease. Haematologica 2008;93:1211-8.
2. Epstein E. Beitrag zurchemie der Gaucherschen krankeit. Biochem Z. 1924;145:398–402.
3. Jain VV, Yelwatkar S. Unusual presentation of adult Gaucher’s disease: A long and difficult road to diagnosis.Indian J Endocr Metab 2011;15:224-6.
4.Nagral A. Gaucher Disease. J Clin Exp Hepatol. 2014 Mar; 4(1): 37–50.
5.Schnabel D, Schroder M, Sandhoff K. Mutation in the sphingolipid activator protein 2 in a patient with a variant of Gaucher disease. FEBS Lett. 1991;284:57–9.
6.Ho M.W., Seck J., Schmidt D. Adult Gaucher's disease: kindred studies and demonstration of a deficiency of acid beta-glucosidase in cultured fibroblasts. Am J Hum Genet. 1972;24:37–45.
7.Giraldo P., Cenarro A., Alfonso P. Chitotriosidase genotype and plasma activity in patients type 1 Gaucher's disease and their relatives (carriers and non carriers) Hematologica. 2001;86:977–984.
8. Svennerholm L., Håkansson G., Lindsten J., Wahlström J., Dreborg S. Prenatal diagnosis of Gaucher disease. Assay of the beta-glucosidase activity in amniotic fluid cells cultivated in two laboratories with different cultivation conditions. Clin Genet. 1981;19:16–22.
9.Di Rocco M, Giona F, Carubbi F, Linari S, Minichilli F, Brady RO, et al. A new severity score index for phenotypic classification and evaluation of responses to treatment in type I Gaucher disease. Haematologica 2008;93:1211-8.
Published
2016-10-03
Issue
Section
Case Report
Authors who publish with this journal agree to the following terms:
- Authors retain copyright and grant the journal right of first publication with the work simultaneously licensed under a Creative Commons Attribution License that allows others to share the work with an acknowledgement of the work's authorship and initial publication in this journal.
- Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the journal's published version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgement of its initial publication in this journal.
- Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See The Effect of Open Access at http://opcit.eprints.org/oacitation-biblio.html).

